INTRODUCTION
Pulmonary arteriovenous malformations (PAVMs) can be associated with a wide spectrum of neurologic manifestations including cerebral infarction, intracranial hemorrhage, and brain abscess due to paradoxical embolization. Approximately 80-90% of PAVMs are found in patients with hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome [1]. HHT is a rare autosomal dominant genetic disorder and vascular dysplasia characterized by telangiectases and arteriovenous malformations (AVMs) in particular locations described in the Cura├¦ao criteria [2]. We report a brain abscess due to idiopathic PAVM in a patient who initially was diagnosed ŌĆśsuspected HHTŌĆÖ but had negative results in genetic testing and no definite family history.
CASE REPORT
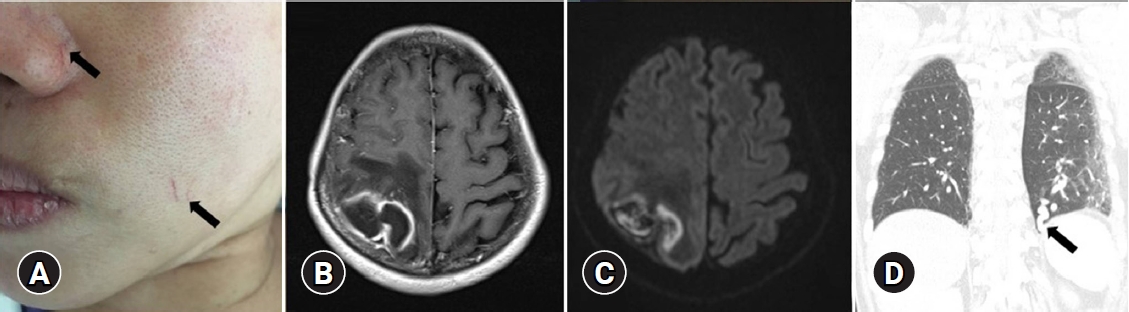
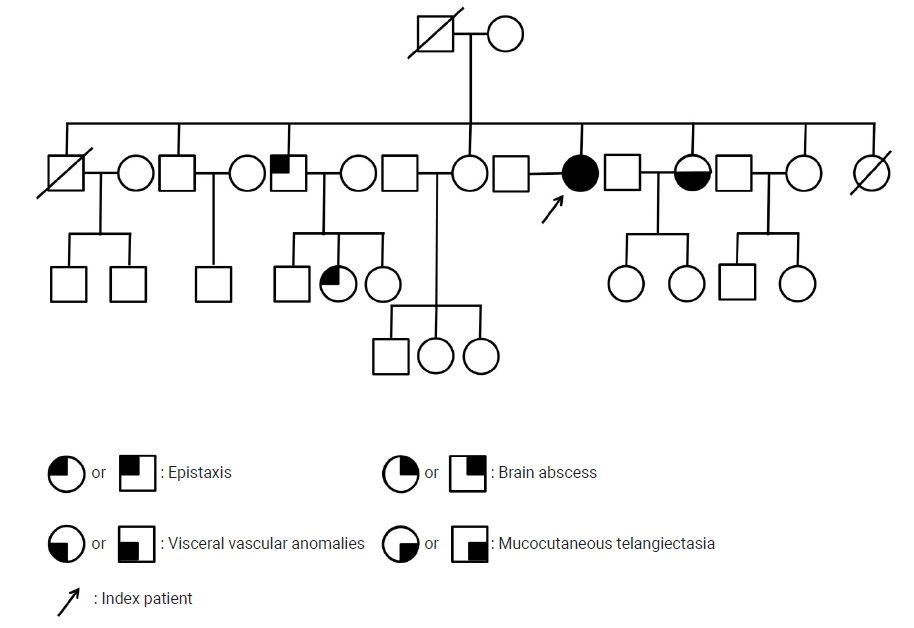
A 53-year-old female presented to the emergency department of Dongguk University Gyeongju Hospital with severe headache and progressive left side weakness for several days. She reported no remarkable past medical history or family history of genetic disorders. Medical examination revealed motor grade III/III strength in the left upper and lower extremities and multiple small telangiectasias on the facial skin (Fig. 1A). Brain magnetic resonance imaging scans revealed a 5.0├Ś3.2 cm ring enhanced mass in the right parietal lobe, with accompanying severe surrounding edema (Fig. 1B, C). The chest X-ray revealed no underlying lung disease, but chest computed tomographic angiography (CTA) showed AVM in the left lower lobe (Fig. 1D). Echocardiography showed normal global left ventricular systolic function; sinusitis and ear infection were also negative. A thorough background check of the patientŌĆÖs family history revealed that one of her brothers and his daughter had frequent nosebleeds, and one sister had been diagnosed with cerebral AVM and facial mucocutaneous telangiectasia (Fig. 2). Based on the Cura├¦ao criteria, the patient was initially diagnosed with suspected HHT, but the genetic analysis for diagnostic confirmation revealed negative findings for ENG and ACVRL1 mutations. The recommended additional mutation analysis of MADH4 and genetic testing for her family was refused by the patient due to financial constraints. However, since there was no certain family history of HHT and also clinically the patient could not be ŌĆśdefinitelyŌĆÖ defined as HHT, we decided to regard the patient as having idiopathic PAVMs. We performed a right osteoplastic craniotomy and abscess removal. Bacteriological cultures obtained intraoperatively were negative, and the patient was treated with intravenous antibiotics (cefotaxime 1 g/8 hr and metronidazole 500 mg/8 hr) for six weeks. At the end of the treatment protocol, there was no headache and complete improvement in hemiparesis with the aid of physical therapy. Imaging showed near-total resolution of the abscess. Following discharge, the patient received pulmonary angiography and embolization at another hospital.
DISCUSSION
PAVMs are characterized by an abnormal vascular connection between an afferent pulmonary artery and one or more efferent pulmonary veins without an interposed capillary bed. The pulmonary capillary bed works as a filter of the bloodstream that removes small thrombi and septic particles. Bypassing the pulmonary capillary bed provides a direct right-to-left shunt that depends on the diameter of the feeding artery [1], allowing the systemic venous blood to bypass gas exchange and pulmonary capillary bed processing. As a consequence, thromboembolic and septic emboli arising in the pulmonary circulation may evoke cerebrovascular disorders, one of which is the formation of a brain abscess.
Approximately 80-95% of PAVMs are associated with HHT, which is also known as the Osler-Weber-Rendu syndrome. HHT is a rare systemic angiodysplasia inherited as an autosomal dominant disorder, characterized by telangiectases and arteriovenous malformations. The widely accepted Cura├¦ao criteria is the standard for diagnosis of HHT, and is based on the most characteristic features of the disease: i) spontaneous and recurrent nosebleeds, ii) mucocutaneous telangiectasia, iii) familial history of HHT in the first-degree relative, and iv) visceral AVM. A diagnosis of ŌĆśdefinite HHTŌĆÖ is reached if a patient exhibits at least three of the four criteria, ŌĆśsuspected HHTŌĆÖ if two criteria are present, and ŌĆśunlikely HHTŌĆÖ for fewer than two criteria [2]. For patients with definite clinical HHT, genetic testing is not required to confirm the diagnosis. The goal of genetic testing for HHT is to identify the causative mutation in a family with clinically confirmed HHT, to establish a diagnosis in relatives of a person with a known causative mutation, and to assist in establishing a diagnosis of HHT in individuals who do not meet the clinical diagnostic criteria. Since more than 80% of all HHT cases are due to mutations in either the endoglin gene (ENG) on chromosome 9 (coding for the endoglin protein) or activin receptor-like kinase gene (ACVRL1) on chromosome 12 (coding for the activin receptor-like kinase 1 protein), the primary recommended genetic test includes analysis of these genes [3,4]. More recently, mutations of the gene called mother against decapentaplegic homolog 4 (MADH4, coding for the SMAD4 protein) have been described in 1-3% of HHT patients who present with a rare syndrome of combined familial juvenile polyposis and HHT. Hence, the HHT Guidelines Working Group recommends that if the complete gene analysis for the ENG and ACVRL1 genes is negative, SMAD4 testing should be considered to identify the causative mutation [5]. However, no matter how remarkable the genetic testing techniques have developed, the final diagnosis of HHT is still based on clinical manifestations because the existing genetic testing is only about 80% sensitive. In the current case, we clinically diagnosed the patient with ŌĆśsuspected HHTŌĆÖ according to the Cura├¦ao criteria; however, the characteristic genetic tests were negative, with no definite family history or any other condition associated with acquired PAVMs. Therefore, the patient was considered to have a brain abscess resulting from idiopathic PAVMs.
The clinical manifestations and complications of idiopathic PAVMs are similar to those associated with HHT and anatomically similar to HHT-related PAVMs, with notable differences of a more significant proportion of solitary PAVMs and a lack of lower lobe predominance [6]. Chest CTA is the gold standard for diagnosis, and transcatheter embolization is the treatment of choice for PAVM. However, because this modality can fail if the PAVM is large or has multiple complex feeding vessels, surgical resection is sometimes necessary for those kinds of patients [7].
CONCLUSION
Here, we report a brain abscess resulting from an idiopathic PAVM in a patient who was initially diagnosed as ŌĆśsuspected HHTŌĆÖ but had negative genetic testing results. We propose that PAVMs should be considered in patients with brain abscess without a known cause. In cases with identifiable PAVMs, it is important to be mindful of the possibility of HHT, and the need for detailed family history and genetic testing of patients with suspected HHT.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print